4月1日,金沙集团1862cc免疫化学研究所白芳课题组在国际学术期刊Journal of the American Chemical Society Au (JACS Au)发表了题为“Accurate characterization of binding kinetics and allosteric mechanisms for the HSP90 chaperone inhibitors using AI-augmented integrative biophysical studies”的研究论文,报道了一种基于多种机器学习算法开发的可准确预测药物-靶标结合动力学的计算模型,应用于肿瘤靶标HSP90开展基于结合动力学与结合亲和力的新型抑制剂的发现,结合蛋白质结构解析技术与基于物理模型的计算生物学技术揭示其变构分子机制。
药物与靶标的结合动力学性质主要包括结合速率常数(Kon)与解离速率常数想(Koff),分别表征了药物与靶标的结合/解离速度与作用时间,是药物药效和安全性的重要指标之一。与最常用的药效指标药物-靶标结合亲和力的预测相比,结合动力学性质与标的构象变化、化合物的物理化学性质等因素相关,计算更为复杂。近年来报道的用于预测结合动力学的计算方法存在计算成本高、耗时长、准确性不足等局限,且缺乏在药物开发中的实际应用。因此,高效和准确的结合动力学预测方法并开展药物设计应用是亟待关注的研究领域。
本项工作基于收集获得的靶向热休克蛋白90αATP结合域(N-HSP90α)的132种抑制剂及其结合动力学实验数据,分别提取化合物结构、三维构象空间结构和抑制剂-靶蛋白结合的复合物结构的特征描述符,分别采用偏最小二乘法(PLS)、极端梯度提升(XGBoost)和支持向量机回归(SVR)等机器学习方法成功构建了基于不同机器学习算法的结合动力学速率常数定量预测模型,如图1。相比已报道的预测算法,该模型为当前最优方法,模型的决定系数和平均绝对误差分别为0.933和0.182。同时,在此基础上本工作明确了影响HSP90抑制剂解离速率常数的关键因素,并应用于新型HSP90抑制剂的发现工作。
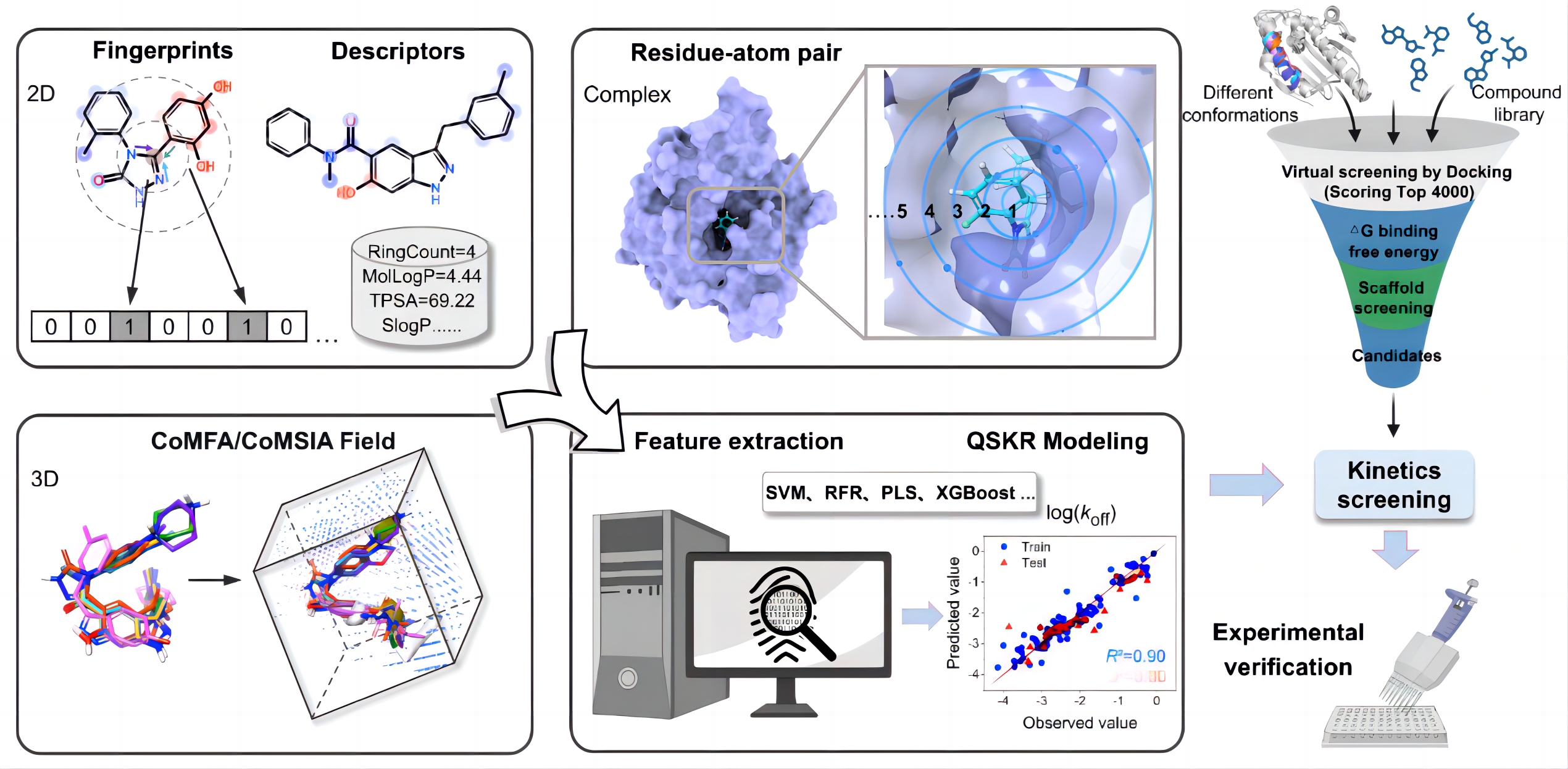
图1. 药物-靶标结合动力学预测模型构建及应用流程示意图。
通过采用基于亲和力的虚拟筛选和基于结合动力学的筛选相结合的策略,发现31个与HSP90具有明显结合信号的苗头化合物,测定了其中16个结合较强的化合物与HSP90的结合动力学数据,实验数据与模型预测结果具有较高的一致性,进一步验证了模型的预测准确性与可靠性。进一步解析了N-HSP90α的单体(apo)结构以及与多个抑制剂分子的复合物结构,发现单体N-HSP90α存在不同构象。不同的配体可以进行构象选择或改变构象平衡,诱导构象重排,从而对结合动力学产生影响。随后通过粗粒度元动力学模拟揭示了单体N-HSP90α的不同构象之间的转变分子机制,如图2。N-HSP90α结构中“lid”(残基102-137)的灵活性是多种构象变化的基础,具有较长滞留时间和诱导螺旋构象能力的抑制剂对N-HSP90α的构象分布具有较为显著的影响。综上,本工作为开发更具成药性的N-HSP90α靶向抑制剂或针对其他靶标的新型药物分子设计提供了新的研究策略和范式。
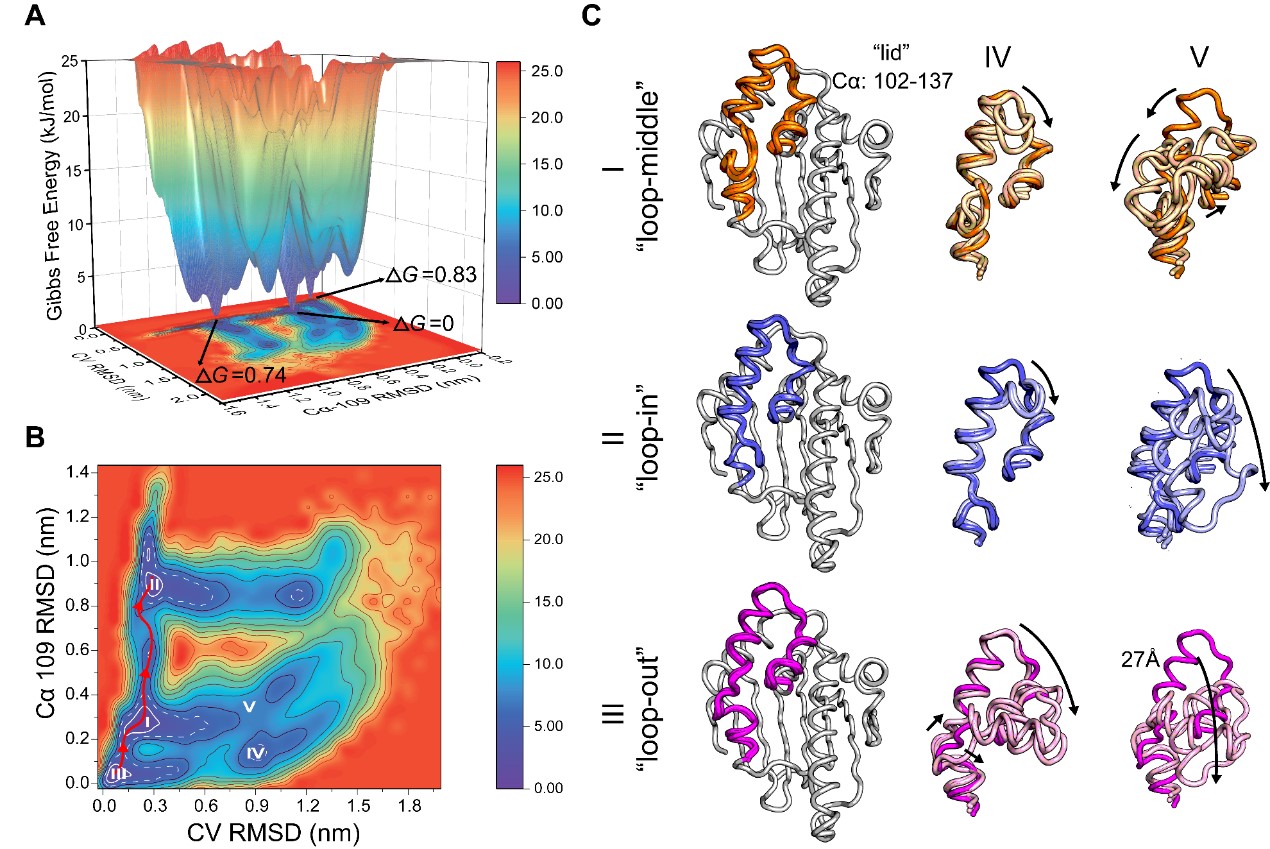
图2. 分子动力学模拟研究N-HSP90α的构象变化过程。
生命科学与技术学院/免疫化学研究所2021级硕士研究生徐超和免疫化学研究所助理研究员张向磊为本文共同第一作者。白芳为通讯作者。金沙集团1862cc为第一完成单位。
 沪公网安备 31011502006855号
沪公网安备 31011502006855号