近日,金沙集团1862cciHuman研究所、生命科学与技术学院水雯箐课题组在《分子与细胞蛋白质组学》(Molecular & Cellular Proteomics)发表了题为“Acquisition and Analysis of DIA-Based Proteomic Data: A Comprehensive Survey in 2023” 的综述论文,对近二十年数据非依赖型采集(data-independent acquisition, DIA)质谱方法做了详细的总结回顾,并提出了对数据分析方法的新分类策略。
DIA于本世纪初开始被用于蛋白质组学研究,近十年随着仪器和计算能力的提升得以飞速发展。由于兼具样品信息覆盖度深和定量重复性高等特点,DIA成为目前自下而上蛋白质组分析中较为常用的一种方法。对DIA质谱数据的采集方式和数据解析方式多种多样,该综述主要涉及多种类型DIA采集方案的特点和归类、不同的DIA数据解析方案、不同数据分析软件实现方式的特点和分类、数据解析中可选的谱图库的构建和优化、可用于测试新数据分析流程的标准数据集。
DIA采集方法的基本特征是在串联质谱中二级质谱采集不依赖于一级质谱所得到的信号,因此任何DIA方法都需要预先设置采集的质量范围。其中最简单的方式是每次都将完整质量范围的离子送入碎裂室和质量分析器(full-scan),但为了降低所采集谱图的复杂程度,一般会将完整质量范围划分为十到上百个窗口再连续分析落入各个质量窗口的离子(windowed)。另外,将DIA和其它采集方法组合可进一步优化在特定场景中所得到的质谱数据的质量(mixed modes)。此三大类中,目前最常用的是使用多个窗口覆盖完整质量范围的方法,已有多种windowed方法可在数据采集方面得到更丰富的信息从而辅助数据解析(如将不同质量窗口相交叠的多种overlapping-window实现)(图1)。
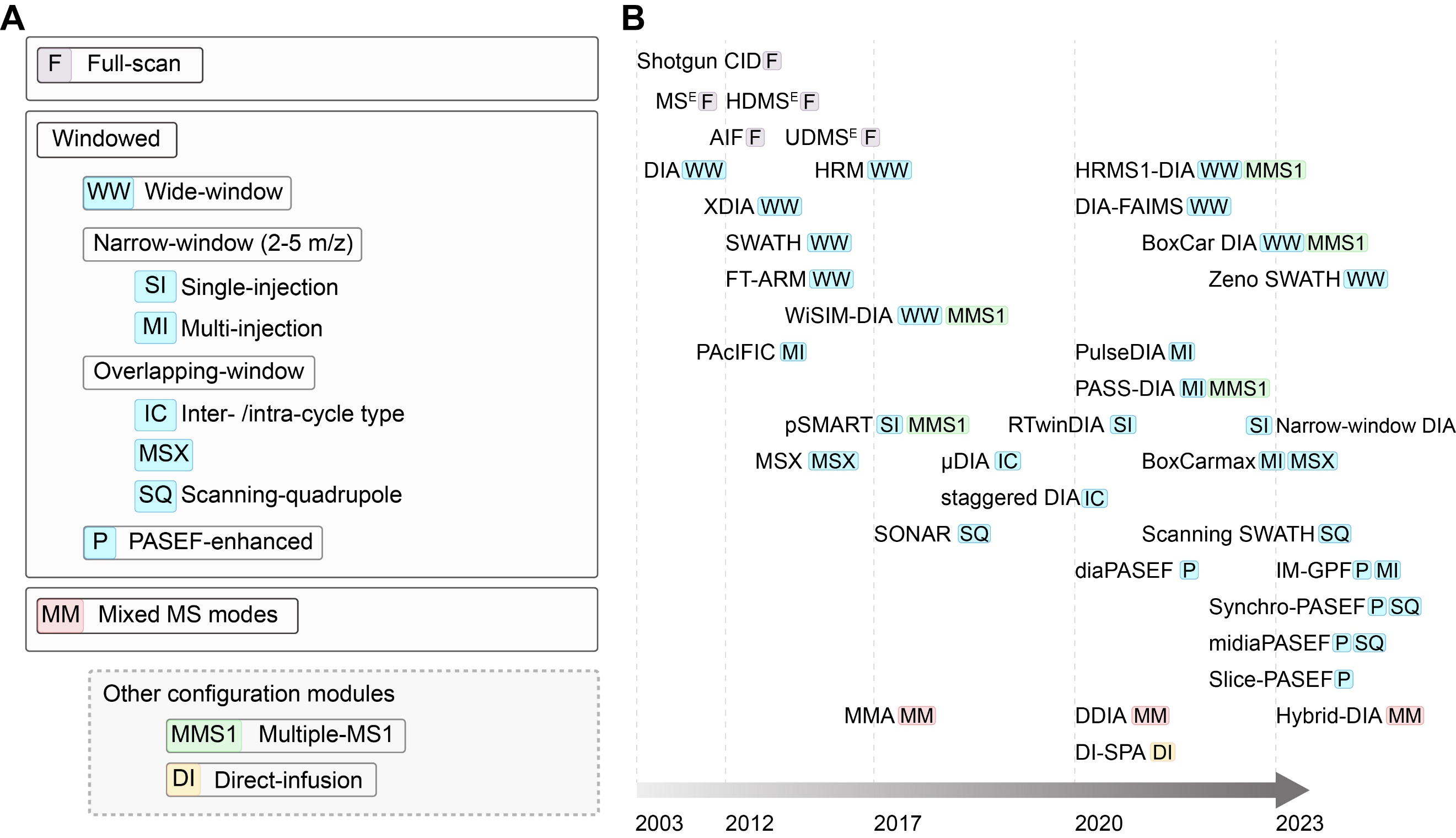
图1:近二十年DIA采集方法的分类(A)和发展时间线(B)
DIA发展历程中的另一个重要问题是数据解析。将一段质量范围的离子同时碎裂并分析将会显著增加每张质谱谱图的复杂程度,其中最自然的方案是根据额外的色谱维度将连续且形状相似的色谱峰归并再匹配简单谱图(即谱图重构)。如果在原始DIA数据上直接解析其所包含的肽段,则需要更稳定的打分系统和可靠的错误发现率评估(包括基于序列或谱图库的检索,以及从头序列分析)。另一方面,每个采集循环的设置保持一致也使得DIA数据可以在损失较少信息的前提下被转换为信号矩阵的表示,矩阵中的特殊模式将会成为数据来源样品的特定信息的证据(即序列无关的方法)(图2)。在以肽段序列鉴定为目的DIA数据分析中,该综述提出两种对相关联的谱图信号进行聚合的方式:第一种以每个色谱时间步的单个质谱谱图做初步匹配,再根据匹配结果选择数个临近谱图中的信号组合;另一种则依次构建每个特定质量的信号的色谱峰再进行匹配、筛选和组合(图2)。
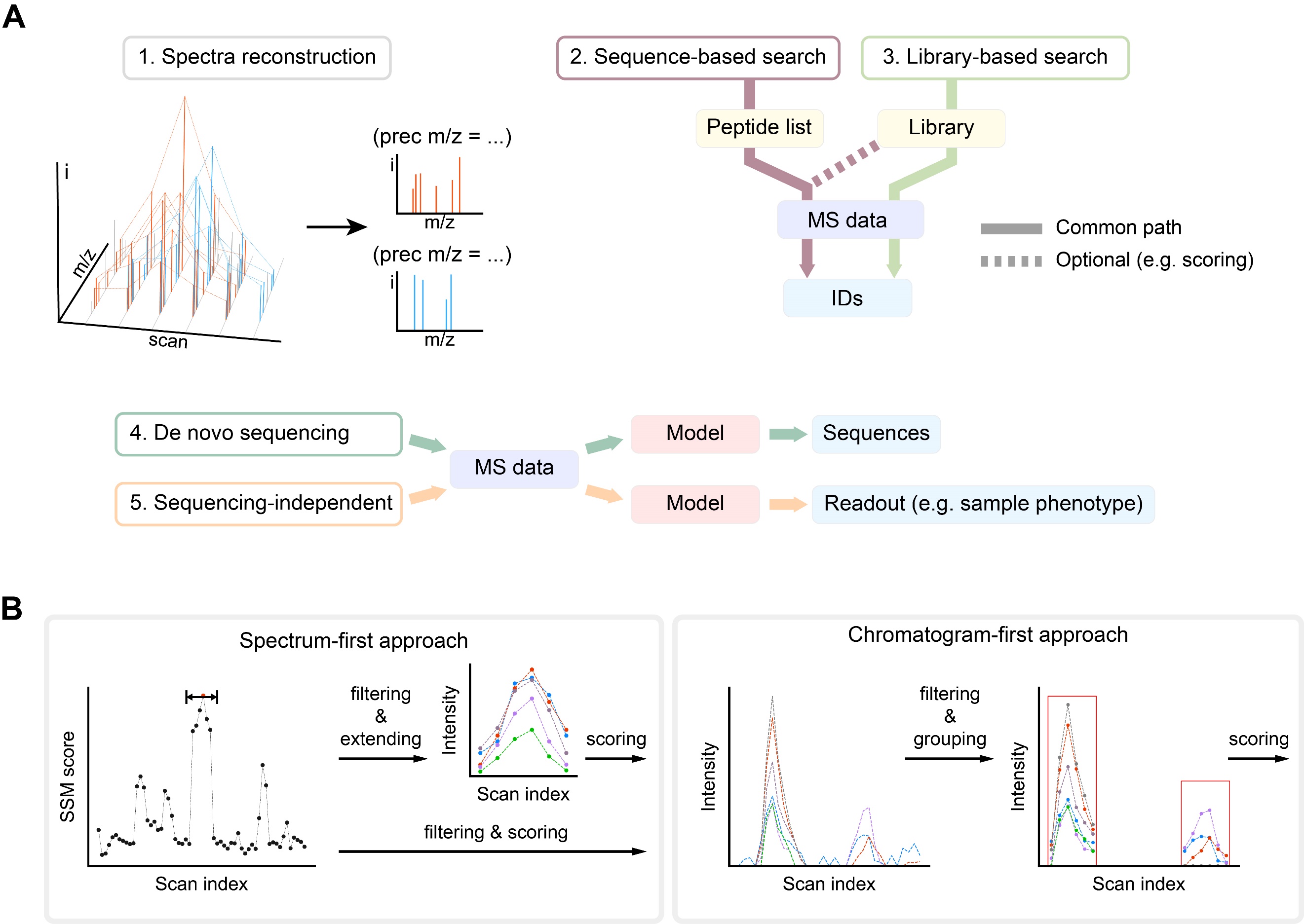
图2. 从DIA数据中获得不同信息的方式(A)和两种组合信号的方式(B)
论文对DIA方法近二十年的发展历程,尤其对不同的采集方法设计和数据解析方式做了详细综述,所提出的新分类方式和对每种方法的特点总结将有助于新DIA方法的开发。
金沙集团1862cciHuman研究所博士后娄容珲为第一作者,娄容珲与生命学院常任副教授、iHuman研究所研究员水雯箐为共同通讯作者。金沙集团1862cc为第一完成单位。
 沪公网安备 31011502006855号
沪公网安备 31011502006855号